Prealignment¶
See the prealignment hydra-genetics module documentation for more details on the softwares. Default hydra-genetics settings/resources are used if no configuration is specified.
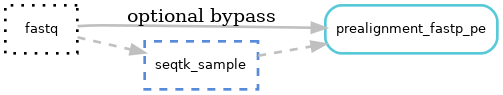
Pipeline output files:¶
Only temporary intermediate files are created.
Downsampling¶
Downsampling of fastq files is performed using seqtk to normalize coverage across samples if needed.
Trimming¶
Trimming of fastq files is performed by fastp v0.20.1.
Configuration¶
Resources
| Options | Value |
|---|---|
| mem_mb | 30720 |
| mem_per_cpu | 6144 |
| threads | 5 |
Merging¶
Merging of fastq files belonging to the same sample are performed by simply concatenating the files with cat.