Fusions in RNA¶
See the fusions hydra-genetics module documentation for more details on the softwares for fusion calling. Default hydra-genetics settings/resources are used if no configuration is specfied.
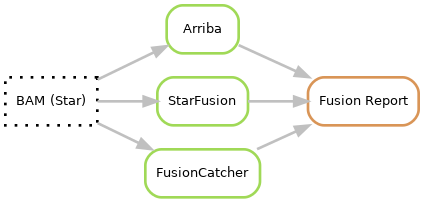
Fusion calling is performed using three different fusion callers; Arriba, Star-Fusion and fusioncatcher. Both Arriba and Star-Fusion uses the Star for alignment but with different settings while fusioncatcher uses its own aligner. After fusion calling the fusions are filtered depending on software and then merged into a fusion report.
Pipeline output files:¶
results/rna/{sample}_{type}/fusion/{sample}_{type}.fusion_report.tsvresults/rna/{sample}_{type}/additional_files/fusion/{sample}_{type}.arriba.fusions.tsvresults/rna/{sample}_{type}/fusion/{sample}_{type}.arriba.fusions.pdfresults/rna/{sample}_{type}/additional_files/fusion/{sample}_{type}.fusioncatcher.fusion_predictions.txtresults/rna/{sample}_{type}/additional_files/fusion/{sample}_{type}.star-fusion.fusion_predictions.tsv
Arriba¶
Alignment with STAR¶
Merged fastq files are aligned with Star v2.7.10a before fusion calling.
Configuration¶
References
- Star genome index - (see references)
Software settings (Recommended options by Arriba)
| Filter | Value |
|---|---|
| --quantMode | GeneCounts |
| --sjdbGTFfile | hg19.refGene.gtf - (see references) |
| --outSAMtype | BAM SortedByCoordinate |
| --chimSegmentMin | 10 |
| --chimOutType | WithinBAM SoftClip |
| --chimJunctionOverhangMin | 10 |
| --chimScoreMin | 1 |
| --chimScoreDropMax | 30 |
| --chimScoreJunctionNonGTAG | 0 |
| --chimScoreSeparation | 1 |
| --alignSJstitchMismatchNmax | 5 -1 5 5 |
| --chimSegmentReadGapMax | 3 |
Cluster resources
| Options | Value |
|---|---|
| mem_mb | 30720 |
| mem_per_cpu | 6144 |
| threads | 5 |
| time | "8:00:00" |
Fusion calling with Arriba¶
Star aligned bam-files are used for fusion calling with Arriba v2.3.0.
Configuration¶
References
- assembly: fasta reference genome
Software settings
| Options | Value | Description |
|---|---|---|
| blacklist | blacklist_hg19_hs37d5_GRCh37_v2.3.0.tsv.gz |
(see references) |
| gtf | hg19.refGene.gtf |
(see references) |
| extra | -p protein_domains_hg19_hs37d5_GRCh37_v2.3.0.gff3 |
(see references) |
| extra | -k known_fusions_hg19_hs37d5_GRCh37_v2.3.0.tsv.gz |
(see references) |
Cluster resources
| Options | Value |
|---|---|
| mem_mb | 30720 |
| mem_per_cpu | 6144 |
| threads | 5 |
| time | "8:00:00" |
Result file¶
results/rna/{sample}_{type}/additional_files/fusion/{sample}_{type}.arriba.fusions.tsv
Fusion images¶
Arriba produces a pdf file containing a figure for every fusion called with a schematic presentation of the exons involved, breakpoints, coverage and directions of the fusion partners in the fusion.
Configuration¶
Software settings
| Options | Value | Description |
|---|---|---|
| cytobands | cytobands_hg19_hs37d5_GRCh37_v2.3.0.tsv |
(see references) |
| gtf | hg19.refGene.gtf |
(see references) |
| protein_domains | protein_domains_hg19_hs37d5_GRCh37_v2.3.0.gff3 |
(see references) |
Result file¶
results/rna/{sample}_{type}/fusion/{sample}_{type}.arriba.fusions.pdf
Star-Fusion¶
Star-Fusion v1.10.1 uses Star to align merged fastq files but do so internally.
Configuration¶
Software settings
| Options | Value | Description |
|---|---|---|
| genome_path: | GRCh37_gencode_v19_CTAT_lib_Mar012021.plug-n-play/ctat_genome_lib_build_dir/ |
(see references) |
| extra | --examine_coding_effect | Add annotation regarding if the fusion is in-frame or not |
Cluster resources
| Options | Value |
|---|---|
| mem_mb | 30720 |
| mem_per_cpu | 6144 |
| threads | 5 |
| time | "8:00:00" |
Result file¶
results/rna/{sample}_{type}/additional_files/fusion/{sample}_{type}.star-fusion.fusion_predictions.tsv
Fusioncatcher¶
Fusioncatcher v1.33 together with reference file package version 102 is used to call fusion from merged fastq files.
Configuration¶
Software settings
| Options | Value | Description |
|---|---|---|
| genome_path | human_v102/ |
(see references) |
Cluster resources
| Options | Value |
|---|---|
| mem_mb | 61440 |
| mem_per_cpu | 6144 |
| threads | 10 |
| time | "16:00:00" |
Result file¶
results/rna/{sample}_{type}/additional_files/fusion/{sample}_{type}.fusioncatcher.fusion_predictions.txt
Fusion filtering and report¶
Fusion candidates from the three fusions callers are collected and filtered with different filtering options for each caller by the in-house script report_fusions.py (rule and config). The remaining fusion calls are then reported in a excel friendly tsv file. Fusions are filtered based on the number of reads cover the breakpoint. However, read pairs spanning the breakpoint are also reported together with total supporting reads as well as other annotations. The settings for respective caller are presented below:
Filter settings¶
| Caller | Option | Value | Description |
|---|---|---|---|
| All callers | Filter fusion when both genes are outside of design | ||
| Arriba | No filters, use Arriba confidence to flag low confidence calls | ||
| Star-Fusion | star_fusion_flag_low_support | 15 | Flags low support when split reads < 15 |
| star_fusion_low_support | 2 | Filters inframe fusions with split read support <= 2 | |
| star_fusion_low_support_inframe | 6 | Filters non-inframe fusions with split read support <= 6 | |
| star_fusion_low_support_fp_genes | 20 | Filters fusions with split read support < 20 if in list of noisy fusions or housekeeping genes (see below) | |
| Fusioncatcher | fusioncather_flag_low_support | 15 | Flags low support when split reads < 15 |
| fusioncather_low_support | 3 | Filters inframe fusions with split read support <= 3 | |
| fusioncather_low_support_inframe | 6 | Filters non-inframe fusions with split read support <= 6 | |
| fusioncather_low_support_fp_genes | 20 | Filters fusions with split read support < 20 if in list of noisy fusions or housekeeping genes (see below) |
In the validation samples the MAML2 gene was falsely called frequently together with a number of different fusion partner genes. These gene combinations as well as the housekeeping have more stringent filtering criteria. The genes affected are listed below:
- MAML2
- FRMPD3, NCOA6, ATXN3, SRP14, KMT2D, CHD1, NFAT5, FOXP2, NUMBL, GLG1, VEZF1, AAK1, NCOR2
- House keeping genes (GAPDH, GUSB, OAZ1, POLR2A)
Configuration¶
Software settings
| Options | Value | Description |
|---|---|---|
| annotation_bed | Twist_RNA_fusionpartners.bed |
Optional file for annotation of fusion partners |
Result file¶
results/rna/{sample}_{type}/fusion/{sample}_{type}.fusion_report.tsv