SNV and INDEL calling, annotation and filtering¶
See the snv_indels hydra-genetics module documentation for more details on the softwares for variant calling, annotation hydra-genetics module for annotation and filtering hydra-genetics module for filtering. Default hydra-genetics settings/resources are used if no configuration is specified.
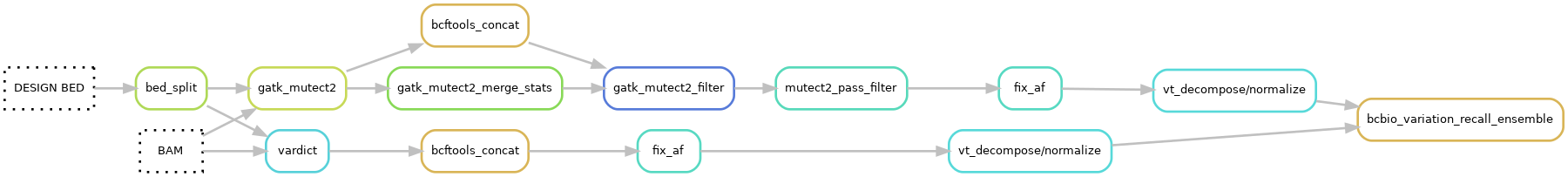
Pipeline output files:¶
results/dna/{sample}_{type}/vcf/{sample}_{type}.annotated.exon_only.filter.hard_filter.codon_snv.vcfresults/dna/{sample}_{type}/vcf/{sample}_{type}.annotated.exon_only.filter.hard_filter.codon_snv.qci.vcfbam_dna/mutect2_indel_bam/{sample}_{type}.bam
SNV and INDEL calling¶
Small variants are called with GATK Mutect2 v4.1.9.0 and Vardict v1.8.3.
GATK Mutect2 variant calling¶
SNVs and INDELs are called by Mutect2 on individual chromosome bamfiles.
Configuration¶
Reference files
- reference fasta genome
- design bed region file (split by bed_split rule into chromosome chunks)
Cluster resources
| Options | Value |
|---|---|
| time | "48:00:00" |
GATK Mutect2 merging¶
The stats file from GATK Mutect2 calling are merged with GATK MergeMutectStats v4.1.9.0 and the vcf files are merged with bcftools concat v1.15.
GATK Mutect2 vcf soft filtering¶
Merged Mutect2 vcf files are softfiltered with GATK FilterMutectCalls v4.1.9.0, which puts filter flags in the vcf FILTER column.
GATK Mutect2 vcf hard filtering¶
Hardfilter Mutect2 vcf files based on the FILTER flags using the in-house script mutect_pass_filter.py (rule). and will only keep variants flagged as:
- PASS
- multiallelic
Vardict variant calling¶
SNVs and INDELs are called by Vardict on individual chromosome bamfiles.
Configuration¶
References
- reference fasta genome
- design bed region file (split by bed_split rule into chromosome chunks)
Software settings
| Options | Value | Description |
|---|---|---|
| bed_columns | -c 1 -S 2 -E 3 -g 4 | bed column definitions |
| extra | -Q 1 | remove reads with 0 mapping quality |
| allele_frequency_threshold | 0.01 | minimal reported allele frequency |
Cluster resources
| Options | Value |
|---|---|
| time | "48:00:00" |
Vardict vcf merging¶
The Vardict vcf files from individual chromosomes are merged with bcftools concat v1.15.
Variant vcf decomposition and normalization¶
Variants called by Vardict and Mutect2 are decomposed by vt decompose follwed by vt decompose_blocksub v2015.11.10. The vcf files are then normalized by vt normalize v2015.11.10.
Variant ensemble¶
Variant vcf files from the two callers are ensembled into one vcf file using bcbio-variation-recall ensemble v0.2.6. All variants from both caller are retained. When both callers call the same variant the INFO and FORMAT data is taken from the Vardict vcf file.
Configuration¶
Software settings
| Options | Value | Description |
|---|---|---|
| support | -n 1 | keep all variant call by at least one caller |
| sort_order | --names vardict, gatk_mutect2 | priority order for retaining variant information |
Annotation¶
The ensembled vcf file is annotated firstly using VEP, followed by artifact annotation and background annotation. See the annotation hydra-genetics module for additional information.
VEP¶
The ensembled vcf file is annotated using VEP v105. VEP adds a pletora of information for each variant which is specified by the configuration flags listed below. Of note are --pick which picks only one representative transcript for each variant, --af_gnomad which adds germline information, and --cache which uses a local copy of the databases for better performance. See VEP options for more information.
Configuration¶
References
- VEP cache including all databases adapted for reference genome GRCh37 and VEP version 105
- Fasta reference genome
Software settings
| Options | Value |
|---|---|
| vep_cache | path_to_vep_cache |
| mode | --offline --cache |
| extra | --assembly GRCh37 --check_existing --pick --sift b --polyphen b --ccds --uniprot --hgvs --symbol --numbers --domains --regulatory --canonical --protein --biotype --uniprot --tsl --appris --gene_phenotype --af --af_1kg --af_gnomad --max_af --pubmed --variant_class |
Resources
| Options | Value |
|---|---|
| mem_mb | 30720 |
| mem_per_cpu | 6144 |
| threads | 5 |
| time | "6:00:00" |
Artifact annotation¶
Identifying artifacts is crucial in a Tumor-only FFPE pipeline such as the GMS560 Twist Solid pipeline. The artifact annotation is performed using the in-house script artifact_annotation.py (rule). The annotation is based on variants called in a number of normal FFPE samples sequenced using the same panel and on the same sequencing machine type as the analysed tumor samples. See references for more information on how the Panel of Normal was created.
Example annotation for one variant added to a vcf file in the INFO field:
| Field | Value | Description |
|---|---|---|
| Artifact | 12,35,36 | Nr of calls made in the PoN using Vardict, Mutect2, and total of samples in the PoN |
| ArtifactMedian | 0.29,0.25 | Median MAF of the calls |
| ArtifactNrSD | 0.58,0.56 | Number of standard deviation between the median allele frequency in the PoN and the call in the variant |
Configuration¶
References
- Panel of Normal with position specific artifact information for each caller and variant type
Hotspot annotation¶
Annotate clinically important variants in the vcf file using the in-house script add_hotspot_annotation.py (rule) and a hotspot list.
Configuration¶
Reference
Background SNV annotation¶
In positions with high background noise it can be hard to distinguish low MAF variants. The background level for all SNVs is therefor added in the vcf file. The background annotation is performed using the in-house script background_annotation.py (rule). It is based on a panel of normal with position specific alternative alleles frequencies obtained from genome VCF files created by GATK Mutect2 v4.1.9.0. See references for more information on how the Panel of Normal was created.
Example annotation for one variant added to a vcf file in the INFO field:
| Field | Value | Description |
|---|---|---|
| PanelMedian | 1.0013 | Median fraction of alternative alleles |
| PositionNrSD | 12.17 | Number of standard deviation between the Median fraction in the PoN and allele frequency of the call in the variant |
Configuration¶
References
- Panel of Normal with position specific background information
Filtering¶
Annotated vcfs are hard filtered first by removing regions outside exons and then filtered by a number of filtering criteria described below. See the filtering hydra-genetics module for additional information. A soft filtered version of the exonic regions is also provided for development and other investigations.
Extract exonic regions¶
Use bcftools filter -R v1.15 to extract variants overlapping exonic regions (including 20 bp padding) defined in a bed file which is a sub bed file of the general design bed file.
Configuration¶
References
- Bed file with exonic regions including 20 bp padding
Hard filter vcf (FFPE)¶
The exonic vcf files for FFPE samples are filtered using the hydra-genetics filtering functionality. The filters are specified in the config file config_hard_filter_uppsala_vep105.yaml and consists of the following filters:
Configuration¶
Software settings
| Filter | Description |
|---|---|
| intron | Hard filter intronic variants |
| vaf | Hard filter variants with low vaf (AF lower than 0.01) |
| artifacts | Hard filter variants found in more than 3 normal samples |
| background | Hard filter position with where backgound distribution overlaps variant (lower than 4 SD from median) |
| germline | Hard filter germline (gnomAD_AF > 0.005) |
| ad | Hard filter variants with few observations (AD lower than 20) |
| ad_hotspot | Hard filter hotspot variants with few observations (AD lower than 10) |
| ad_TERT | Hard filter TERT variants with few observations (AD lower than 4) |
Hard filter vcf (ctDNA)¶
The exonic vcf files for ctDNA samples are filtered using the hydra-genetics filtering functionality. The filters are specified in the config file config_hard_filter_umi_vep105.yaml and consists of the following filters:
Configuration¶
Software settings
| Filter | Description | Criteria |
|---|---|---|
| intron | Hard filter intronic variants | Not splice, not MET/TERT, not COSMIC |
| noisy_gene | Hard filter variants in noisy genes | MUC6, CDC27, KMT2B, KMT2C, KMT2D, HLA-A, HLA-B, HLA-C |
| artifacts_SNVs | Hard filter SNVs found in 3 or more normal samples | Artifact > 2 and ArtifactNrSD < 6 |
| artifacts_INDELs | Hard filter INDELs found in 3 or more normal samples | Artifact > 2 and ArtifactNrSD < 6 |
| background | Hard filter variants with AF not over background noise | PositionNrSD < 6 |
| germline | Hard filter germline | gnomAD_AF > 0.005 |
| af_snv | Hard filter SNV variants with low vaf | AF < 0.003 or AF > 0.997 |
| ad_snv | Hard filter SNV variants with low ad | AD < 10 |
| af_snv_hotspot | Hard filter SNV variants with low vaf (hotspot) | AF < 0.0025 or AF > 0.9975 |
| ad_snv_hotspot | Hard filter SNV variants with low ad (hotspot) | AD < 6 |
| af_insertion | Hard filter Insertion variants with low vaf | AF < 0.005 or AF > 0.995 |
| ad_insertion | Hard filter Insertion variants with low ad | AD < 10 |
| af_deletion | Hard filter Deletion variants with low vaf | AF < 0.005 or AF > 0.995 |
| ad_deletion | Hard filter Deletion variants with low ad | AD < 10 |
| af_complex | Hard filter complex variants with low vaf | AF < 0.009 or AF > 0.991 |
| ad_complex | Hard filter complex variants with low ad | AD < 20 |
| af_substitution | Hard filter substitution variants with low vaf | AF < 0.006 or AF > 0.994 |
| ad_substitution | Hard filter substitution variants with low ad | AD < 15 |
| ald | Hard filter variants with skewed distribution between strands | ALD < 3 |
| sbf | Hard filter variants with skewed distribution between strands | SBF < 0.01 |
| pmean | Hard filter variants found only in start of reads | PMEAN < 15.0 |
| callers | Hard filter variants only called by mutect2 | CALLERS = gatk_mutect2 |
Combine SNVs in the same codon¶
Two or more variants affecting the same codon can have different clinical implications when considered individually compared to in combination. This is because the combined variant could end up coding for a different amino acid then the when only looking at the variant individually. Variants within the same codon are therefore combined and added to the vcf file using the in-house script add_multi_snv_in_codon.py (rule). Codon information is based on the VEP annotation. Annotation information is taken from the variant with the highest allele frequency. After adding the combined variants the vcf is sorted and annotated again.
Configuration¶
Software settings
| Options | Value | Description |
|---|---|---|
| af_limit | 0.00 | No lower limit for allele frequency |
| artifact_limit | 10000 | Allow any number of observations (10000) in the PoN as they are already filtered |
Result file¶
results/dna/{sample}_{type}/vcf/{sample}_{type}.annotated.exon_only.filter.hard_filter.codon_snv.vcf
QCI AF correction of vcf¶
The clinical interpretation tool QCI calculates allele frequency from the AD FORMAT field instead of using the AF FORMAT field supplied by the callers. This has shown to be wrong especially for INDELs. The AD field is therefore corrected so that the allele frequency based on the AD field corresponds to the AF field. This correction of the vcf file is performed by an the in-house script fix_vcf_ad_for_qci.py (rule and config).
Result file¶
results/dna/{sample}_{type}/vcf/{sample}_{type}.annotated.exon_only.filter.hard_filter.codon_snv.qci.vcf
GATK Mutect2 variant bam file¶
When GATK Mutect2 finds INDEL candidates it realignes reads in this regions and outputs a realigned bam-file covering these INDEL regions. This makes it possible to inspect INDELs called by Mutect2 in IGV. As Mutect2 runs on individual chromosomes these bam-files are then merged, sorted and indexed before.
Result file¶
bam_dna/mutect2_indel_bam/{sample}_{type}.bam