Overview of the pipeline¶
Here is a brief overview of the entire pipeline. For details see subsections and the hydra-genetics documentation.
DNA (FFPE)¶
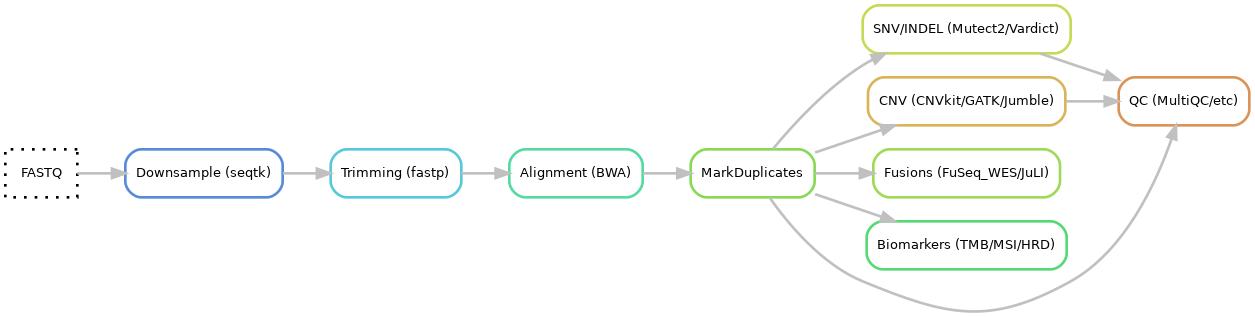
- Input files: fastq
- Downsample: seqtk
- Trimming using fastp
- Alignment using BWA-mem
- Mark duplicates using Picard
- SNV and INDEL
6.1 Calling using Mutect2 and Vardict
6.2 Annotation using VEP and hydra-genetics annotation module 6.3 Filtering using bcftools and hydra-genetics filtering module - CNV
7.1 Calling using CNVkit, GATK CNV and Jumble 7.2 Merging using SVDB 7.3 Annotation using SVDB and hydra-genetics
7.4 Filtering using hydra-genetics
7.5 CNV html report using hydra-genetics filtering module - Fusion calling using FuSeq_WES and JuLI
- Biomarkers
9.1 TMB using hydra-genetics biomarker module 9.2 MSI score using MSIsensor-Pro
9.3 HRD using CNVkit/ScarHRD and Jumble - QC
10.1 QC measures from Samtools, Picard, FastQC, GATK
10.2 MultiQC hmtl report
10.3 Hotspot coverage report
10.4 Sample mixup check
DNA (ctDNA)¶
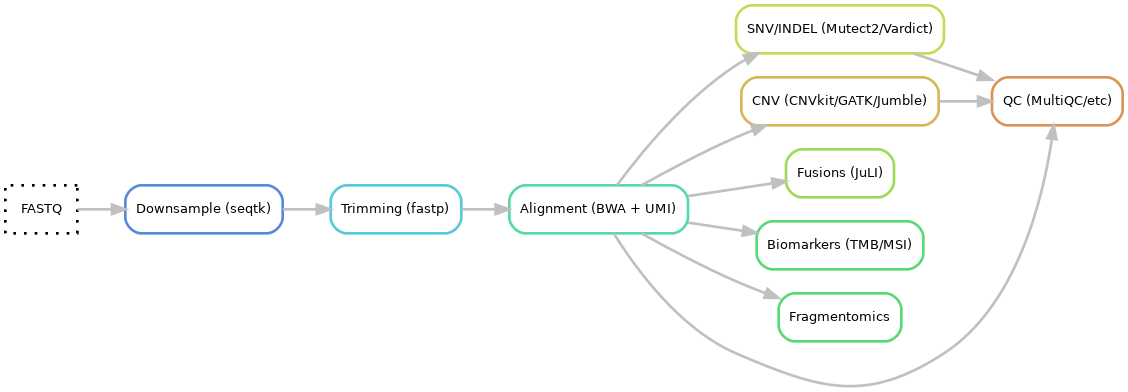
- Input files: fastq
- Downsample: seqtk
- Trimming using fastp
- Alignment using BWA-mem and UMI consensus calling
- SNV and INDEL
5.1 Calling using Mutect2 and Vardict
5.2 Annotation using VEP and hydra-genetics annotation module 5.3 Filtering using bcftools and hydra-genetics filtering module - CNV
6.1 Calling using CNVkit, GATK CNV and Jumble 6.2 Merging using SVDB 6.3 Annotation using SVDB and hydra-genetics
6.4 Filtering using hydra-genetics
6.5 CNV html report using hydra-genetics filtering module - Fusion calling using JuLI
- Biomarkers
8.1 TMB using hydra-genetics biomarker module 8.2 MSI score using MSIsensor-Pro 8.3 HRD using Jumble - Fragmentomics using FinaleToolkit
- QC
10.1 QC measures from Samtools, Picard, FastQC, GATK
10.2 MultiQC hmtl report
10.3 Hotspot coverage report
RNA¶
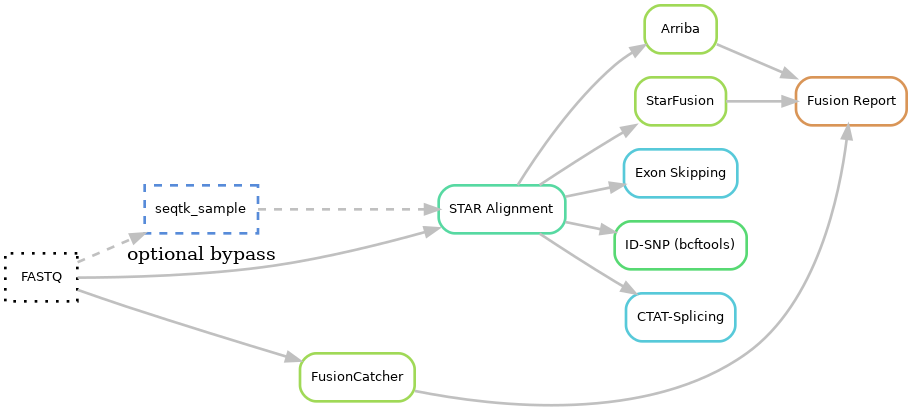
- Input files: fastq
- Downsample: seqtk
- Alignment using Star
- Fusions
4.1 Fusion calling using Arriba, StarFusion, FusionCatcher
4.2 Filtering and report using in-house script
4.3 Fusion images using Arriba - Exon skipping using in-house script and ctat-splicing
- ID-SNP calling using bcftools
- QC
7.1 QC measures from Samtools, Picard, FastQC, Mosdepth
7.2 MultiQC hmtl report
7.3 House keeping gene coverage
7.4 Sample mixup check